5.1 数据折叠 EMP_collapse
数据折叠是指对原始的assay进行合并以生成新的assay。EMP_collapse可以根据rowdata或coldata中的指定数据进行折叠。在折叠过程中,可以选择计算平均值、中位数、求和、最大值或最小值作为合并后的结果。
5.1.1 根据重复的coldata合并assay
EMP_collapse 可以将assay按照coldata进行折叠。
🏷️示例:按照coldata的Group列合并assay。
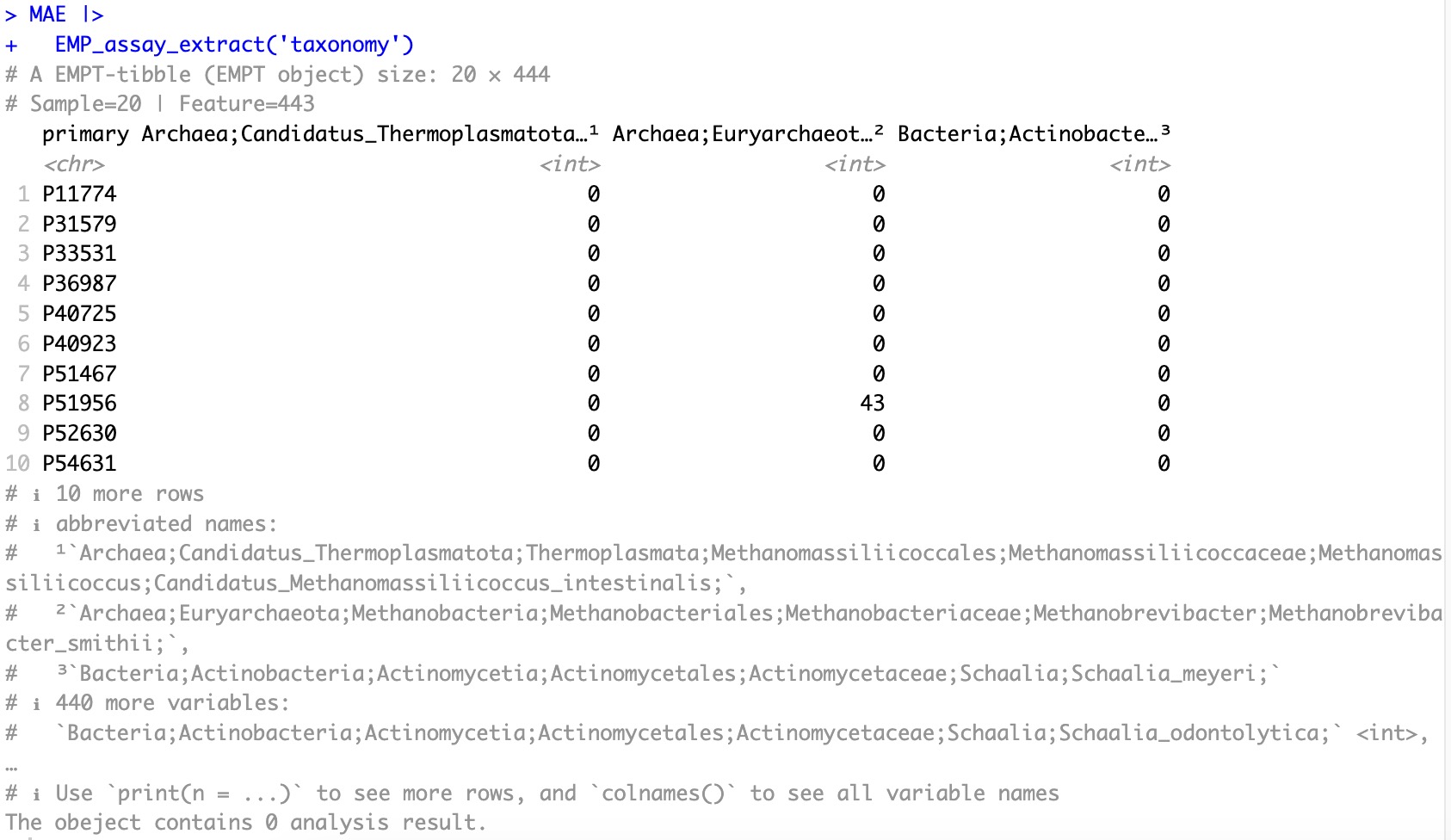
折叠前的assay:
MAE |>
EMP_assay_extract(experiment='taxonomy')
折叠后的assay:
提取组学项目taxonomy的assay,参数estimate_group指定按照coldata的Group列折叠,参数collapse_by指定按照col(coldata)折叠,参数collapse_sep指定使用+分隔符来合并该组学项目的coldata中发生折叠的字符串单元格。
MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='col',estimate_group = 'Group',
method = 'mean',collapse_sep = '+')

折叠后的coldata:
注意:
尽管数据折叠操作本质上是将
尽管数据折叠操作本质上是将
assay进行折叠,但就任何组学项目而言,assay、coldata和rowdata是从建立MAE对象时就绑定、联系在一起的。因此,当assay根据coldata进行折叠时,coldata也会进行相应折叠。如果coldata的数据属于连续型变量,则和assay使用相同的方法进行计算;如果coldata的数据属于字符串变量,则使用collapse_sep指定的符号进行合并。如果coldata的数据属于逻辑变量,则使用collapse_boolean进行合并。
MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='col',estimate_group = 'Group',
method = 'mean',collapse_sep = '+') |>
EMP_coldata_extract()
5.1.2 根据重复的rowdata合并assay
EMP_collapse 可以将assay按照rowdata进行折叠。
🏷️示例1: 按照rowdata的Class列合并assay。
在微生物组学中,输入MAE对象内的微生物物种数据通常为最高分类级别的数据(例如:种或株级别)。EMP_collapse可以通过指定参数estimate_group,将assay的feature按照rowdata内较高的分类级别快速折叠为较低分类级别(例如:门或属级别)。
注意:
①将assay按照rowdata折叠后,assay的
②微生物物种注释折叠过程中可能出现全注释与单注释的冲突问题,请查看微生物数据完整注释问题。
③微生物物种注释折叠过程中,本模块会自动检测上述冲突问题,并提示用户选择全注释或者单注释。由于流程中模块默认进行自动缓存,如果需要重新选择,可以设置
①将assay按照rowdata折叠后,assay的
feature发生改变,但sample是不改变的。在本示例中,原本为species级别的feature被折叠为Class级别。②微生物物种注释折叠过程中可能出现全注释与单注释的冲突问题,请查看微生物数据完整注释问题。
③微生物物种注释折叠过程中,本模块会自动检测上述冲突问题,并提示用户选择全注释或者单注释。由于流程中模块默认进行自动缓存,如果需要重新选择,可以设置
use_cached = FALSE,然后再次运行即可。MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='row',estimate_group = 'Class',
method = 'sum',collapse_sep = '+')

🏷️示例2:按照rowdata的特定列合并assay。
注意:
①在代谢组学项目中,代谢物通常有多种注释方式,我们可以快速按照rowdata中代谢物的注释级别对assay进行数据折叠。
②用于折叠的feature必须存在于rowdata中(例如,组学项目geno_ko的assay无法按照rowdata中的Class列进行折叠)。用户可以使用模块
①在代谢组学项目中,代谢物通常有多种注释方式,我们可以快速按照rowdata中代谢物的注释级别对assay进行数据折叠。
②用于折叠的feature必须存在于rowdata中(例如,组学项目geno_ko的assay无法按照rowdata中的Class列进行折叠)。用户可以使用模块
EMP_rowdata_extract查看可用于合并的feature。
折叠前的assay:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')

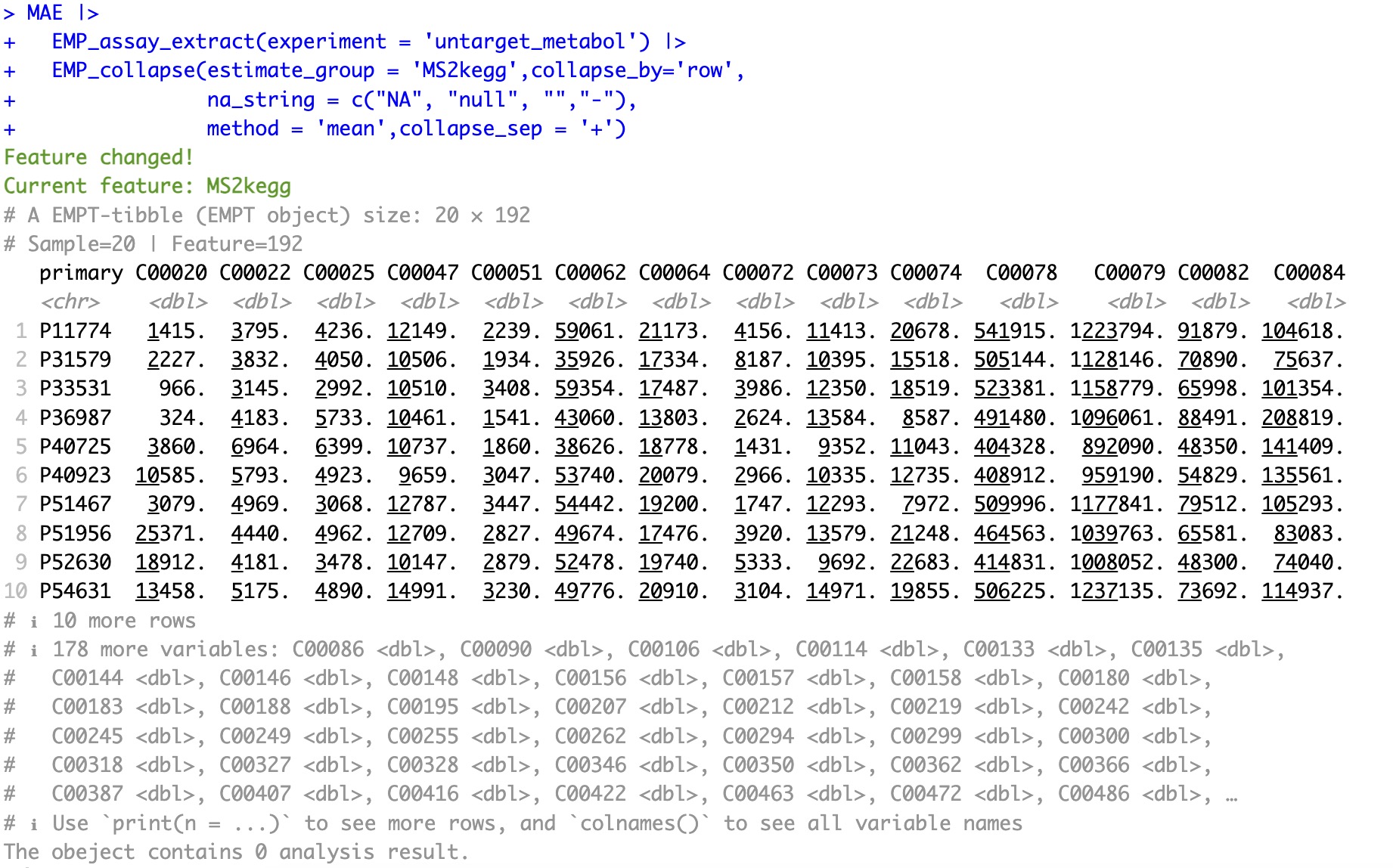
折叠后的assay:按照kegg二级代谢产物折叠后的assay。
注意:
在一些代谢组学原始数据中,经常出现NA、 null、 - 等符号(均表缺失值),使用参数
在一些代谢组学原始数据中,经常出现NA、 null、 - 等符号(均表缺失值),使用参数
na_string可以指定这些字符串为缺失值。在数据折叠时,如果在指定的用于合并的feature中出现缺失值,则该行数据将会被忽略。
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol') |>
EMP_collapse(estimate_group = 'MS2kegg',collapse_by='row',
na_string = c("NA", "null", "","-"),
method = 'mean',collapse_sep = '+')